ファイルが大きくなりすぎたので分割しました。最新の記事はこちらに掲載しています。
「添加回収試験」はどの程度当てになるか?(2003/4/26)
圃場散布で残留した農薬と添加された農薬は、どう違うか?(2003/4/13)
分析ラボ新人が何より先に覚えるべき五か条(2003/4/6)
穀類・豆類などを「水で膨潤」するのは必要か?(2003/4/2)
農作物中の残留農薬は、まずどんな溶媒で抽出すべきか?(2003/4/2)
農薬の分析法をバリデートするとき、また、ルーチンでの精度管理を行うとき、主に「添加回収試験」が行われている。既知の濃度の農薬を添加した試料を分析し、分析値が添加量にどれだけ近いか(つまり、回収率がどの程度100%に近いか)を指標にして信頼性を評価する。これは、最も普通に行われている方法だ。
ところで、「添加回収試験」は、それほど信頼できるものなのだろうか?Horwitzらが2000年に発表した論文(文献1)は、私が日頃漠然と持っていた疑問を、はっきりした言葉で表現してくれた気がした。
Horwitzらは、FAPASRのシリーズ19として行われた第1ラウンド〜第5ラウンド(1997年7月〜1999年1月)のデータを集計して考察している。(FAPASR=ファーパスについてはこちらで解説した。)FAPASRでは、主催者が添加した農薬を、各参加機関が分析して定量値を報告する。また、試験品と同じ種類の食品を使って内部で添加回収試験を行った機関は、その結果も報告する。つまり、各機関は、添加量が既知(known)か未知(unknown)かという点を除いて全く同じ試験をする。その場合、各機関が出す分析値のセットは、たとえばこんな分布になると予想される。(knownとunknownのデータが、100%を中心に、どちらも同じくらいばらつくとすれば。)
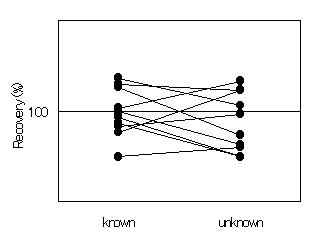
ところが、Horwitzらが解析した結果は、こんな風だった。
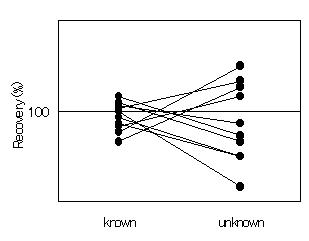
添加量がわからない回収試験よりも、添加量を分析者が知っている回収試験のほうが明らかに分析値のバラツキが少なく、値は100%に近かった。また、Horwitzらはこのようにも書いている。「分析者みずからが行う添加回収試験の値と外部で添加された農薬の試験結果との間には、驚くことに何の相関関係もなかった。」「相関係数は+1になるべきところ、18のデータセットで-0.5〜+0.7に分布しており、平均は+0.1であった。回帰直線は45度になるべきなのに(こうなるはずなのに)
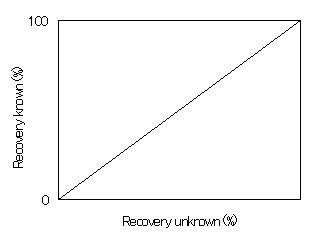
実際には、内部精度評価の添加回収率100%付近の水平な線になった。(つまりこういう風になった。)
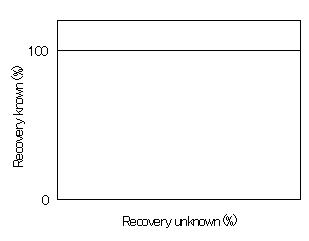
いったい、なぜこんなことが起こるのか?その理由としてHorwitzらは「既知の添加量での試験の場合、多くの分析者が(意識的か無意識的かはともかく)自分の結果を加算または減算して100%に近づくように手を加えたことを示している」と推定している。
「100%に近づくよう結果に手を加える」とは、どういうことか?これは、実際に農薬を分析したことがある人なら、必ず思い当たるふしがあるはずだ。
食品の抽出液は、精製してもかなりの妨害物質を含んでいる。林のようなピークが並ぶクロマトグラムから農薬を探す場合も多い。添加回収率が100%から大きくはずれていたら、分析者はもう一度クロマトグラムを見返して、「妨害ピークを測り込んでいないか」とか「自動処理で引かれたベースラインがおかしくないか」とか「近傍の別のピークを読んでいないか」とか、確認するだろう。その結果、データが手直しされて、より100%に近い値になる場合がある。添加量がわかっていなければ、このようなフィードバックは行われないから、多数機関のデータで全体としてknownとunknownの結果が違ってくるのもうなずける。
ただし、このHorwitzらによる解析のみから「添加回収試験には意味がない」と言ってしまうのは早計だろう。FAPASRに参加する機関が使用する試験法は、多くの場合、既にバリデートされている。つまり、もともと100%に近い値が得られる試験法であろう。そういうデータ範囲の中だけで論じると、100%近辺での相対的に小さなバラツキを過大に重視することになりかねない。非常に問題のある試験法(誰がやっても100%と掛け離れた回収率になるような)を発見するためには、やはり添加回収試験は有効と考えられる。
それでもHorwitzらの論文は、分析者自身が添加量を知っている回収試験の限界を浮き彫りにしていると言える。実際の農薬分析は、「100%」と最初から答えのわかっているテストのようなものではない。バイアスを排除した精度管理や分析法バリデーションを行うためには、分析者が予め添加量を知ることのない仕組みを整えて回収試験を行うべきだろう。
文献1 Horwitz, W., Wood, R., Relationship of (known) control values to (unknown) test values in proficiency studies of pesticide residues. J. AOAC Int., 83, 399-406 (2000)
分析法の精度管理のための添加回収試験。これはごく普通に行われていることだ。今ちょっと考えてみたいのは、分析法のバリデーションのための添加回収試験。これもごく普通に行われている。しかし、例えば添加回収で90%の結果が得られる試験法なら、実用的にも問題ないと言えるのだろうか。そんな疑問を持って調べてみた。
圃場で農産物に散布される農薬には粘土鉱物や界面活性剤等の助剤が加えられており、これらの助剤が薬剤の作物に対する付着の様式に影響を及ぼす。また、根から吸収された薬剤、葉表面の気孔から浸透した薬剤は、植物表面に単に付着した薬剤とは植物組織との結合様式が異なると考えられる。
一方、実験室内で添加される農薬は、多くの場合有機溶媒に溶解されており、農産物の表面に直接ふりかけたとしても、その結合様式は圃場で散布された場合とはまったく異なるであろうことは容易に想像できる。農産物と接触する時間も非常に短い。Kleinは既に1958年の報告(文献1)で、「このような方法は農産物由来のバックグラウンドを確かめるためのものであって、分析法全体の評価には使えない」と述べている。
AOAC(米国公的分析化学者協会)は1950〜1970年代に、実際に圃場で農薬を散布された農産物を用いて抽出法を精査している。抽出効率の評価は、放射性同位元素でラベルした農薬を用いたり、抽出効率がほぼ100%であることが確かめられているソクスレー抽出と比較する等の方法で行われている。その結果、水分含量の少ない試料に水を加える必要性や、最初の抽出を酢酸エチルで行ったらどうなるかが明らかにされた。
つまり、添加回収実験を分析法のバリデーションに使う際には、少なくとも最初の抽出法については圃場散布農薬でバリデートされているかどうかを確認しなければならないということだ。一方、いったん抽出溶媒に移行した農薬に関しては、残留していたものと添加されたものとで挙動に本質的な違いがあるとは考えにくい。つまり、最初の抽出以降の試験法検討においては、添加回収のデータだけでOKだろう。実際、それを当然の前提として書かれている論文しか読んだことがない。
文献1 Klein, A.K., Peport on extraction procedures for chloro-organic pesticides, J. AOAC, 41, 551-555 (1958)
就職・人事異動の季節です。分析ラボにも新人が入って来ます。個々の操作は追々覚えてもらえばいいとして、とりあえず、失敗が実害に結びつく「これだけはするな!」というNG集がないものでしょうか。既にあるのかもしれませんが、私なりのものを作ってみました。
キャピラリーカラムの液層がはがれてMSに吸い込まれてしまいます。カラムがダメになり、イオン源も汚れます。このような失敗が起こり得るのは、GC/MSの立ち上げの手順を間違えた場合、バルブ操作を誤ってキャリアーを止めてしまった場合、真空状態で長期間放置されているGC/MSに接続したボンベが空になってしまったのに気づかなかった場合等が考えられます。
(真空系は常に真空にしておくのがメンテナンス上望ましいため、使用頻度の低いGC/MSでも真空を維持することが多い。)
空気が入るとカラムは劣化します。カラムだけでなく、流路に空気が入るのも、追い出すのが難しくて手こずる場合があります。HPLCを動かしている間はずっと、移動相の残量に気を配りましょう。
サンプル瓶を並べたらホッとしてしまいがちですが、洗浄液が少なくなっていないか、確認を忘れてはなりません。もし忘れたら、当然シリンジの洗浄が無効になってデータに影響が出ます。それだけでなく、空気を吸い上げる動作を繰り返すことにより、シリンジの密閉度が劣化します。HPLCの洗浄液は大容量ですから頻繁に補充する必要がありませんが、それだからこそ、いつの間にか忘れてしまい、忘れた頃に切れてしまう可能性があります。
「シリンジをダメにする」は、新人がラボに金銭的損害を与える失敗として最も可能性が高いものだと思います。(新人でなくてもよくやる。)針はだいたい形状記憶合金でできていますが、一度曲げてしまうと、外見上は元に戻っても曲がり癖がつき、さらに失敗しやすくなります。特にGCのシリンジは細い上、注入口セプタムの材質も固いですから、必ず垂直に構えて慎重に差し込みましょう。
HPLCのシリンジは太く、注入口は柔らかいですが、それにもかかわらずHPLCのシリンジを曲げてしまった新人は、GCの場合以上に深く反省してください。
酸性・アルカリ性・または緩衝液を移動相にして分析した後は、まず系内に水を流し、次にアセトニトリルかメタノールを流して、その状態で装置を止めてから帰りましょう。HPLCカラム内の粒子表面は、とてもデリケートに作られています。特にアルカリ性が強い移動相を使った場合ほど、そのままの状態で放置するのはカラムをいためます。
キリよく五か条にまとめてみました。ラボによっては不用なものもあるでしょう。(例えば、すべてのクロマトグラフをオートサンプラーだけで注入するように設定している場合、4は不要。)個々の事情に応じて削ったり付け加えたりして、新人に申し渡してはいかがでしょうか。
余談:最近は、安全装置付きで失敗そのものが起こらない設計になっている機器が多いですが、そういう行き届いた仕組みが多くなるほど機器はブラックボックス化し、愛着がわきにくくなるような気がします。例えばFIDが自動点火になっているGCでは、炎が消えて水素ガスが漏れる危険は減りますが、FIDがどんな仕組みで分析対象を検出するのか、ほとんど実感できません。
一昔前のFIDは、水素ガスを多めに流しながら火花タイプのライターで点火し、ポッと音がしたらレコーダーの反応を見ながら炎を消さないようにガスを絞って所定の条件に合わせるというものでした。そういう実感を持ちながら使っていると、FIDに異常が起こった時、自然にまずベースラインに着目します。「ノイズがある=炎はついている」「完全にフラット=炎が消えている」と、具体的にイメージしながら異常の原因を探っていけます。
しかしまあ、世の中全体が複雑化していますから、仕方ないことなのでしょう。機器と仲良くしすぎていては、仕事が進みません。あまりこだわらずに、サービスマンを呼びましょう。
「膨潤」という言葉はどの程度一般的なのだろうか。農薬を分析する人以外には、あまりなじみのない言葉だろう。これは、穀類・豆類などを分析するときに、試料と同量や二倍量の水を加えて、ある程度の時間放置すること。公定法では多くの場合「2時間」ということになっている。
2時間というのは長い。朝から分析を始めても、2時間待ったらお昼になってしまいそうだ。なぜこんな操作が必要なのか?と誰でも考えるだろう。
そこで調べてみたところ、やはり残念ながら、水分含量の少ない試料を水を用いずに有機溶媒のみで抽出して良好な結果が得られたという報告は皆無だった。
Bertuzziら(文献1)は、水分7%のビートパルプ中のディルドリンを0〜40%の水を加えたアセトニトリルで抽出した結果、35%含水アセトンでの抽出効率が最高であったとしている。水を加えないアセトニトリルでは、35%含水アセトンによる場合の17%程度しか抽出されなかった。また、試料を水で10分間膨潤してからアセトニトリル及び含水アセトニトリルで抽出する方法を、ビートパルプ中のディルドリン及びピーナツ干し草中のp,p'-DDT、o,p'-DDT、p,p'-DDEで比較した結果、共に本質的な差はなかったとしている。
Burkeら(文献2)は中程度の水分含量の試料について検討している。圃場でTDEを散布したケールを収穫して自然乾燥し、様々な水分含量の試料を作製した。そしてアセトニトリル・35%含水アセトニトリル及びソクスレーの3とおりの抽出法で分析した。その結果、水分が82%含まれる試料では3法の間に検出量の差はなかったが、水分含量が少なくなるにつれてアセトニトリルのみでは抽出効率が落ち、水分26%で抽出率44%、水分13%で同36%となった。Burkeらは、水分が約75%未満の試料については35%含水アセトニトリルで抽出すべきだとしている。
その後Burkeら(文献3)はさらに対象農薬を広げて検討した。生鮮ケール及びコラード(ケールの一種)中のディルドリン、ダイアジノン、エンドスルファン、メトキシクロール、パラチオン、TDEをアセトニトリルで抽出した場合92〜104%回収された。(ソクスレー抽出との比較。)しかし乾燥して水分含量を15〜75%に減らしたケール中のメトキシクロール、エンドスルファン、ディルドリンの場合は、35%含水アセトニトリルで水分含量に関わらず96〜108%抽出されたのに対し、アセトニトリルでは水分含量が少ない試料ほど抽出率が低く、最低20〜30%しか抽出されなかった。残留濃度が低い場合ほど、より抽出効率が低い傾向があるとBurkeらは述べている。
以上、有機塩素系、有機リン系の農薬について、水を用いずにアセトニトリルのみで抽出する方法は抽出効率が低いことが、1970年代までに確認されている。「残留農薬分析マニュアル」には、「乾燥試料は、それ自身の水分含量が極端に低いため、水と有機溶媒の協力によって抽出される。この目的のために水・アセトニトリルの使用がいくつかの研究から支持されている。」と書かれている。(文献4)
水が必要だということは明快にわかった。しかし、私には消えない疑問がある。一つは、なぜ「2時間膨潤」なのかということだ。FDAのように35%含水アセトニトリルを使えば、そんな待ち時間は不用ではないか。二つめは、公定法(「食品、添加物等の規格基準」の試験法)に「穀類に水を加えずに有機溶媒のみで抽出」という方法がいくつも含まれていること。アルジカルブ、エチオフェンカルブ等試験法の抹茶及びホップとか、イプロジオン試験法の穀類、豆類、種実類、抹茶とか。これらの公定法は、どんな経緯で作られたのだろう。
上記の疑問は在職中に解決しておきたかったのだが時間切れになってしまった。ご存知の方がおられたら、ぜひ教えてほしい。メール
文献1 Bertuzzi, P.F., Kamps, L., Miles, C.I., Burke, J.A., Extraction of chlorinated pesticide residues from nonfatty samples of low moisture content, J. AOAC, 50, 623-627 (1967)
文献2 Burke, J.A., Porter, M.L., Note on the effect of sample moisture content on extraction of TDE from kale, J. AOAC, 50, 1260-1262 (1967)
文献3 Burke, J.A., Porter, M.L., Young, S.J.V., Evaluation of two extraction procedures for pesticide residues resulting from foliar application and root absorption, J. AOAC, 54, 142-146 (1971)
文献4 米国食品医薬品局編、PAM日本語版編集委員会訳「FDA残留農薬分析マニュアル」(中央法規、2000)p.93
農作物から農薬を抽出する第一段階で使う溶媒は何を選択すべきか?現在では多くの場合、アセトンまたはアセトニトリルということになっている。米国FDA「残留農薬分析マニュアル」(文献1)には、「高水分含有試料から残留化学物質を抽出するために水と混和する溶媒を用いる必要性について、長い時間をかけて確立されてきた」と書かれている。そして、アセトンかアセトニトリルで抽出したら、次には水と混和しない酢酸エチルやヘキサンなどに転溶して、液々分配で水溶性不純物を除いたり、ミニカラム精製を行ったりする。
しかし、農薬分析に一とおり慣れてくると、誰でもあれこれと手抜き法を思いつくものだ。いっそのこと最初から酢酸エチルで抽出してしまえばどうだろう?一次抽出から転溶までには減圧濃縮やケイソウ土カラム、アセトニトリルなら塩析といったステップが必要だが、こういう面倒なことを省けるのではないだろうか?実際、農薬登録保留基準の試験法では、ベノミル等の極性の高い農薬について酢酸エチルでの抽出を採用している。一般的にはダメだと教科書には書いてあるわけだが、いったいどの程度ダメなのか?
という疑問から、「残留農薬分析マニュアル」の元文献をたどってみた。
実は、果実及び野菜中の有機リン系農薬を直接酢酸エチルで抽出する方法は、1970年代初頭にはFDAの試験室でルーチン的に使用されていたとの記述がある(文献2)。つまり、この方法は農薬分析の初期においてはむしろメジャーな方法だったのかもしれない。まずシンプルな方法から始まったのは自然なことだ。
Wattsらは、ビーンプランツ中のカルバリル、マラチオン、フォスファミドン、また、ケール中のアジンフォスメチル、パラチオン、マラチオンについて、酢酸エチル抽出・アセトニトリル抽出・ソクスレー抽出の3法を比較し、どの方法でも抽出効率に差はなく良好であったと結論づけている(文献2)。
しかしBurkeらは別の結果を得ている(文献2)。Burkeらはケール中のパラチオンとダイアジノンの場合、酢酸エチル抽出ではアセトニトリル抽出の73〜85%程度の抽出効率であったとしている。また、ケール中のp,p'-TDE(有機塩素系)の場合は、アセトニトリル抽出ではソクスレー抽出の99%が抽出されたのに対し、酢酸エチルでは3回抽出でも86%であったとしている。
これらの報告は相互に矛盾する点もあるが、果実・野菜を試料とする場合、有機リン系、カルバメート系、有機塩素系の農薬はアセトニトリルではほぼ100%抽出され、酢酸エチルでは70〜80%以上抽出されると言えそうだ。
このような根拠によって、農薬を正確に定量するためには水と混和する溶媒を使用しなければならないという合意が作られてきたのだろう。しかし、酢酸エチルでも70〜80%以上抽出されるなら、スクリーニング試験に使うことはできるのではないだろうか?分析法はシンプルなほどコストを抑えられるし実験廃棄物も少ない。まずはシンプルな方法で多数の食品を調べて、農薬が見つかったものだけ正確な定量をすればいいのではないか?
という着想から、国衛研での最後の仕事では、実際にこの方法を使ってみた。結果はまもなく公表の予定だ。
(なお、文中引用した文献のデータは、いずれも添加回収でなく圃場散布農薬での値。)
文献1 米国食品医薬品局編、PAM日本語版編集委員会訳「FDA残留農薬分析マニュアル」(中央法規、2000)p.93
文献2 Watts, R.R., Extraction efficiency study---examination of three procedures for extracting 14C-labeled and unlabeled residues of organophosphorus pesticides and carbaryl from bean leaves and kale, J. AOAC, 54, 953-958 (1971)
文献3 Burke, J.A., Porter, M.L., A study of the effectiveness of some extraction procedures for pesticide residues in vegetables, J. AOAC, 49, 1157-1162 (1966)
管理者:津村ゆかり yukari.tsumura@nifty.com